
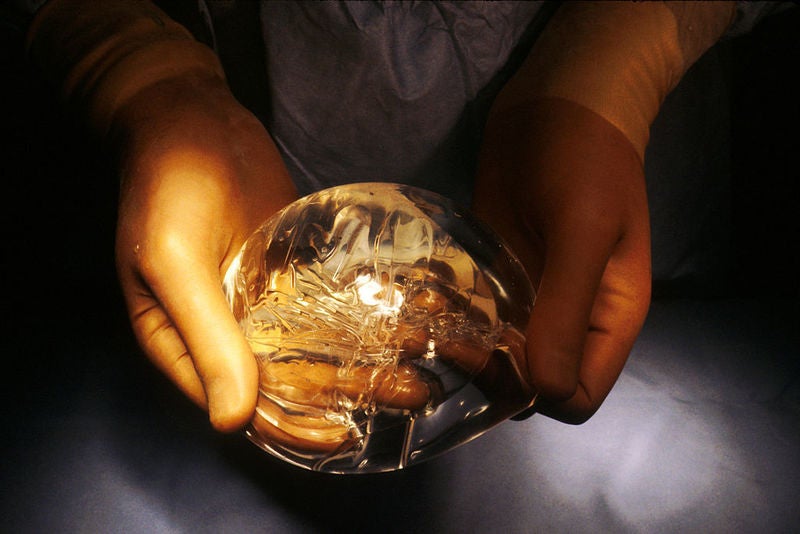
The US Food and Drug Administration (FDA) has sent warning letters to a Johnson & Johnson unit, Mentor Worldwide, and Sientra for failure to comply with its post-approval study requirements for breast implants.
The letters warned about the companies’ post-approval studies evaluating the long-term safety and risks of the silicone gel-filled implants.
Go deeper with GlobalData

Discover B2B Marketing That Performs
Combine business intelligence and editorial excellence to reach engaged professionals across 36 leading media platforms.
In the letter to Mentor Worldwide, the agency highlighted multiple deficiencies in the company’s post-approval study for the MemoryShape breast implant approved in 2013.
The FDA noted that the company failed to enrol the required number of subjects in the study, had poor follow-up rates with participants, along with significant data inconsistencies such as poor patient accounting.
Based on the review of various interim study reports submitted by Mentor, the FDA initially found that progress on the post-approval study appeared adequate.
However, the agency said that Mentor was advised of concerns about patient enrolment, follow-up rates and data inconsistencies, and the company failed to address these issues.
Meanwhile, Sientra has been warned over poor follow-up rates in its post-approval study. The agency said that the company reported a follow-up rate of 61%, which is below target.
The FDA added that it notified Sientra about the inadequate study progress due to low follow-up rates, but the company failed to address the concern.
FDA commissioner Scott Gottlieb said: “We’re issuing these warning letters based on the manufacturers’ low recruitment, poor data, and low follow-up rates in their required post-approval studies.
“Post-approval studies, along with other surveillance tools such as adverse event reports, registries, and scientific literature, allow the FDA to help ensure the safety of medical devices and protect patients.”
The FDA requested the companies to respond within 15 working days with details about how the noted violations will be corrected. The agency may take action if the companies do not comply.
Earlier this month, the agency announced a focus on evaluating materials used in medical devices and potential biological response that patients may have to certain materials in implantable or insertable devices.
