
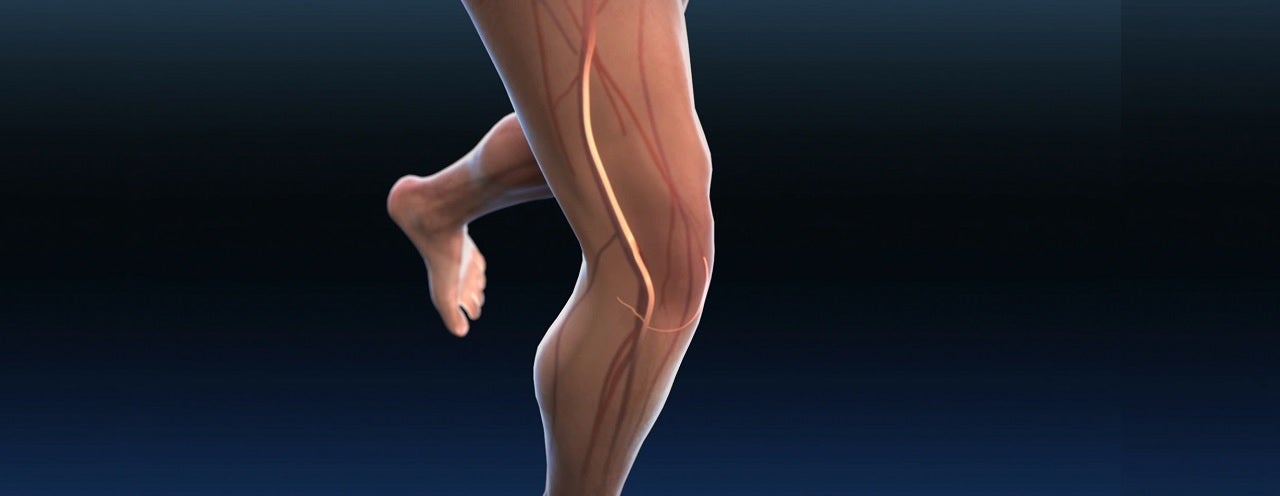
Medtronic has received the Food and Drug Administration (FDA) approval for the Abre venous self-expanding stent system.
The device is intended for use in the iliofemoral veins in patients with symptomatic iliofemoral venous outflow obstruction, also known as deep venous obstruction.
Go deeper with GlobalData

Discover B2B Marketing That Performs
Combine business intelligence and editorial excellence to reach engaged professionals across 36 leading media platforms.
The deep venous obstruction is caused when the veins in the deep venous system become obstructed, blocked, or compressed, leading to a restricted blood flow to the heart.
If left untreated, the condition may cause leg discomfort and pain in patients, affecting their mobility and quality of life.
Swelling in the leg, skin changes, leg ulcers, and pain are among the symptoms of the disease.
The condition may also lead to severe complications, including blood clots that affect lungs, a clot in the leg called deep vein thrombosis (DVT), or the formation of fibrotic tissue or scarring caused by a chronic DVT.
Medtronic Cardiac and Vascular Group endoVenous business vice-president and general manager Carolyn Sleeth said: “With Abre, our goal was to create a dedicated venous stent that combined a balance of the key characteristics necessary to treat patients with a broad spectrum of deep venous obstruction.
“We are excited to bring Abre to the US market, which we believe will provide both physicians and patients with a new option backed by clinical evidence to treat this disease safely and effectively.”
The FDA approval is based on the 12-month outcomes reported by ABRE clinical study, which evaluated the safety and effectiveness of the investigational Abre stent in 200 patients with iliofemoral venous outflow obstruction.
The study achieved its 12-month primary effectiveness endpoint with an overall primary patency rate of 88.0%.
Abre secured CE Mark approval in April of 2017. It can also be used in the iliofemoral veins for treatment of symptomatic venous outflow obstruction.
In a separate development, the FDA has granted to 510(k) clearance to Abiomed for its all-in-one, compact cardiopulmonary bypass system, Abiomed Breethe OXY-1 System.
The system offers cardiopulmonary bypass support for patients whose lungs are unable to generate necessary end organ oxygenation. The clearance enables the use of the device for pumping, oxygenating, and removing carbon dioxide from blood during cardiopulmonary bypass for approximately six hours.
Additionally, it offers oxygenation to patients suffering from cardiogenic shock or respiratory failure related ARDS, H1N1, SARS, or Covid-19, the company noted.
Meanwhile, in another major development, Foldax has secured FDA approval to expand the US clinical study of the Tria surgical aortic heart valve.
The company plans to start the next stage of enrolment next month.
