


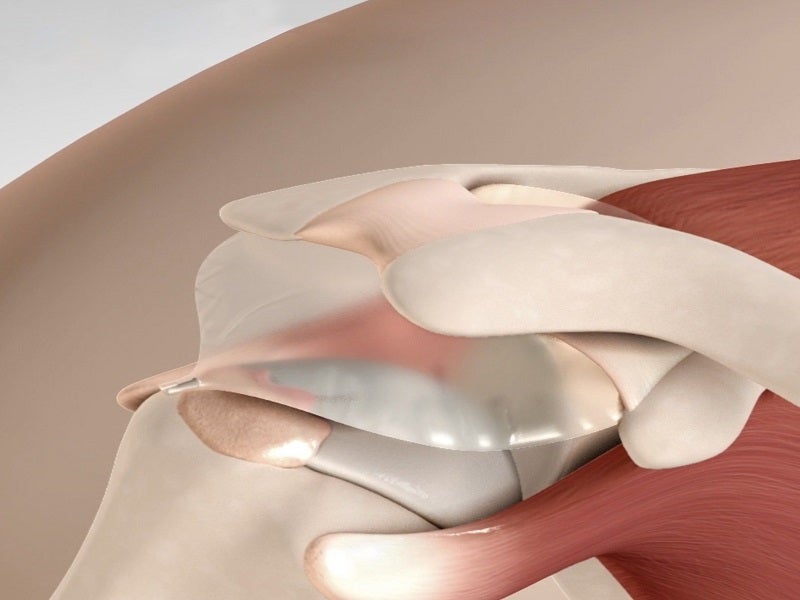

The InSpace implant is a first-of-its-kind biodegradable subacromial balloon spacer designed for the arthroscopic treatment of massive irreparable rotator cuff tears (MIRCTs).
Originally developed by Israel-based orthopaedic devices firm OrthoSpace, the InSpace implant was added to Stryker’s portfolio after Stryker acquired OrthoSpace in March 2019.
Recommended Buyers Guides
The InSpace balloon is a breakthrough solution designed to restore the subacromial space. It provides a shorter, less invasive option for surgeons in shoulder care, facilitating continuous, clinically substantial improvements in the function and symptoms of the patient’s shoulder.
The patients eligible for the implant are those with MIRCT, aged 65 and older, with weakness in shoulder rotation, muscle atrophy or ruptured rotator cuff tendons.
Regulatory approvals and availability
The device received clearance from the US Food and Drug Administration (FDA) in July 2021. The inaugural InSpace implant procedure was conducted in December 2021.
InSpace holds CE Mark certification and is marketed in Europe, Russia, Israel and several Latin American and Asian countries.
The device has a successful clinical track record spanning a decade, with 29,000 balloons implanted internationally. Before its introduction to the US market, Level I research was conducted across North America.
Challenges with MIRCTs
MIRCTs are one of the most common causes of shoulder dysfunction. MIRTCs are considered irreparable according to the preprocedural magnetic resonance imaging (MRI) or intra-operative assessment, which makes it physically and biologically challenging for the surgeon to repair them.
Current techniques for MIRCT treatments may require lengthy and frustrating patient rehabilitation processes.
The average rate of rotator cuff re-tear following surgery is around 20% to 40%, while the failure rates of massive tears can reach 70%. An option to significantly reduce the patient’s pain has been highly sought-after by surgeons.
InSpace balloon implant details
InSpace is a minimally-invasive, single-use, biodegradable implantable balloon system which includes an introducer and a pre-shaped balloon spacer made of a biodegradable copolymer.
The copolymer brings together the durability of poly-L-lactide, constituting 70% of the blend, and the flexibility of e-caprolactone, making up the remaining 30% of the composition. It is not toxic, does not elicit inflammation and biodegrades over 12 months.
The implant is designed to restore the subacromial space without the need for sutures, fixation devices or any kind of discomfort. It can improve shoulder mobility and function.
The balloon is inserted using a cylinder-shaped insertion tube, which is withdrawn once the spacer is placed into the subacromial space. It is implanted, either arthroscopically or via a five-step mini-open surgery, between the acromion and the humeral head or rotator cuff.
The device creates a space between the bone structures, allowing the humeral head to glide smoothly and frictionlessly against the acromion.
The InSpace system is delivered sterile and is free of latex and phthalates.
Benefits of the InSpace balloon implant
The InSpace balloon implant is indicated for the treatment of full-thickness MIRCTs due to trauma or degradation with mild to moderate glenohumeral osteoarthritis in patients aged 65 years or older and whose clinical conditions may improve by a shorter surgical time treatment compared with the partial rotator cuff repair.
The procedure is streamlined and the deployment of the implant from introduction to sealing and retraction takes an average four minutes.
The implant is a game-changing treatment for the arthroscopic repair of large irreparable rotator cuff injuries. The efficient treatment also delivers long-term patient benefits.
Clinical trials on the InSpace balloon implant
The FDA’s approval of the InSpace balloon implant was based on the outcome of a prospective, single-blinded, multi-centre, randomised, controlled, pivotal clinical trial conducted to assess the safety and efficacy of the implant compared with the surgical partial repair of full-thickness MIRCT.
A total of 184 patients were randomised to receive either arthroscopic InSpace (sub-acromial tissue spacer system) implantation or arthroscopic partial repair of the rotator cuff.
In the primary safety outcome, InSpace was found to be non-inferior to the partial repair and demonstrated a high rate of clinical success (87.8%), compared with partial repair (88.1%), at 24 months.
InSpace offered a substantial advantage of reducing the operating time to 44 minutes, whereas the partial repair took 71 minutes.
Results showed similar rates of subsequent secondary surgical intervention during the study and clinically substantial improvements in shoulder function and symptoms after two years. No serious adverse device effects were observed in patients at any point in the study.
The results demonstrated that the InSpace implant is an appropriate alternative to partial repair in patients with MIRCTs.
It also revealed notable patient benefits including early functional recovery and pain relief combined with a shorter operative time.