
On 5 April 2017, two new Medical Devices Regulations (MDR) on medical devices were adopted, and they entered into force on 25 May 2017.
These replace the existing Directives:
- Regulation (EU) 2017/745 (MDR – Medical Device Regulation) repealing Council Directives 90/385/EEC and 93/42/EEC (MDD).
- Regulation (EU) 2017/746 (IVDR – In Vitro Diagnostics Regulation) repealing Directive 98/79/EC and Commission Decision 2010/227/EU.
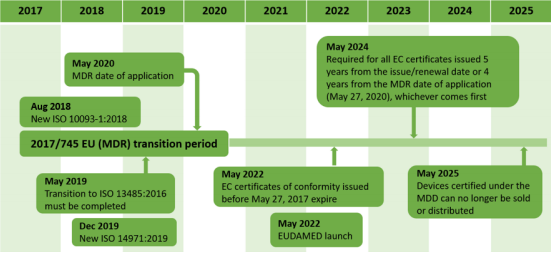
The new rules will only apply after a transitional period: MDR (26 May 2020) and IVDR (26 May 2022). The transition from MDD to MDR, which is, in fact, a transition from a directive to a regulation, provoked an avalanche of changes in the regulatory area, such as major revisions of relevant standards (e.g. ISO 14971:2019 and ISO 10993- 1:2018) and also created a long period (5 years) where 2 different conformity assessment procedures are applied. As can be seen from the following figure, from May 2020 to May 2025, the same products will be placed into the EU market under both MDD and MDR, which constitutes a paradox, considering the totally different requirements of each legislation.

The new regulations aim to increase the safety and effectiveness of medical devices in the EU market, not only at the pre-market phase but throughout the life-cycle of the product. The MDR and IVDR have emerged as a response to the technical and scientific developments that are quickly shaping the medical device industry.
Driving towards the last months before the MDR and IVDR implementation, the Medical Devices community (including the Notified Bodies – NBs) have faced and are still facing many expected and unexpected issues. Most of the EC estimations regarding the smooth transition from the MDD to the MDR were not confirmed, leaving the manufacturers into uncharted territories, trying to catch the train of transition without the proper resources. Moreover, most of the NBs stopped accepting new clients for the MDD before the end of 2019, in order to focus on their MDR designation. Until now, the pace of NBs designations for the MDR is considerably slower than expected (only 9 so far at the beginning of 2020), which will negatively impact the CE marking process for at least the first two years (2020-2021) after entry into force. As a result, it is expected that a significant number of NBs may not be renotified, or may not be notified for the same scope, which could force some medical device manufacturers to change NBs.
On the other side, EC remains optimistic for the transition and continues working on exhaustive pace, publishing guidelines and trying to provide the missing elements which are mentioned in the MDR. However, all this huge amount of information provided at the last minute rather confuses and overloads the manufacturers and the NBs, making the transition to the MDR a Sisyphean effort.
The regulations feature several major changes and efforts. A quick summary of the key changes, aiming to aid organizations in their plans to transition to the new regulations, is presented below.
- Broadened definitions of regulated Medical Devices: Introduction of nonmedical and cosmetic devices, as well as “reusable surgical instruments category” (Class IR).
- Specified Role of Economic Operators (manufacturers, distributors, importers, suppliers, subcontractors, assemblers, and EU Authorized Representatives).
- Person Responsible for Regulatory Compliance (similar to “Qualified Person”).
- Classification rules: Changed from 18 (MDD) to 22 (MDR).
- Unique Device Identification (UDI) and implant cards (for implantable devices).
- Vigilance and Post-market Surveillance: PMS plan and report, Periodic Safety Update Report – PSUR for Class IIa, IIb and III medical devices and EUDAMED (European database for Medical Devices) are introduced.
- Summary of Safety and Clinical Performance: For implantable devices and for class III devices the manufacturer shall draw up a summary of safety and clinical performance and make it available via EUDAMED.
- Clinical Evaluation and Clinical Investigation: “Expert panels” for Class IIb implantable and Class III. Clinical investigations and Post Market Clinical Follow-up (PMCF) will be required in more cases.
- General Safety and Performance Requirements (GSPR): MDR continues the approach of MDD’s “Essential Requirements – ER” with the GSPRs, also in Annex I expanded in 23 sections (see Figure below).
- Common Specifications (CS): Provide a means of complying with GSPR.

Conclusion
While moving towards May 2020, the Medical Device community is still trying to adjust to the “new world” of the MDR. All new changes have to be tested into real cases, in order for the NBs to get into action and gain the required experience through implementation. It is clear that the community is far from being ready and the first estimations indicate that the transition will not be as smooth as it was expected.
There are a lot of pending issues that have not been resolved from the EC, such as the expert panel for clinical evaluation or EUDAMED while the low number of designated NBs makes the task even harder.
Implementation of the UDI, significant changes to technical files and the quality system, as well as the generation of additional clinical evidence for devices currently on the market are some of the biggest burdens manufacturers will face in the next few years. Project management is crucial for all manufacturers, acting proactively, including gap analysis for the transition and proper resources management.
